




IECE Transactions on Advanced Computing and Systems
ISSN: 3067-7157 (Online)
Email: [email protected]

Positively-sense RNA viruses belonging to the family Coronaviridae (CoVs) are responsible for causing various infections in humans, animals, and birds [1]. There are four generations in this family, namely alphacoronavirus, betacoronavirus, deltacoronavirus, and gamammoronavirus [2]. Two of the most notorious conditions in the beta virus species are (SARS-CoV) [3] and (MERS-CoV) [4], which have contaminated thousands of people throughout the world in the last two decades. With the current drug discovery system, newly discovered medicines will take several years to reach the market [25]. Rapidly, due to the recent outbreak of atypical pneumonia (dubbed COVID-19) caused by the new Coronavirus (SARS-CoV-2, Figure 1 illustrates the structural representation of SARS-CoV-2) in Wuhan, China, the world is in the same situation as the previous wave [1, 5]. As per the latest updates from the WHO, the global medical community has not yet identified any exceptional individuals in medicine for effectively eradicating COVID-19 [6]. People dealing with hydroxychloroquine (HCQS) as a therapy for this condition expressed surprise and excitement [7]. However, medical practitioners hesitate to disseminate the data with the same zeal because it has yet to be shown beneficial. When dealt with, it was revealed that remedisivir is good in the United States, but it is not yet available to treat patients with COVID-19 infection. It is considered a safe option until an important study has been carried out simultaneously [8]. The latest news has shown clinical trials in COVID-19 patients in New York heartburn drugs [9]. In summary, there is currently no established treatment to combat the COVID-19 virus. However, the utilization of artificial intelligence (AI) tools in the medical field has the potential to facilitate a feasible cure [9]. DL models [10] were recently accepted as a breakthrough, providing a new chance to make computer decisions based on pharmaceutics. Small molecules that move on a protein target can be identified when analyzing protein constructions using structural-oriented medicine design methods [26]. So, the proficient research capacity here presents a new ingredient. If biochemists validate it as an effective solution, it will help humanity survive these difficult times. We will design a deep learning model to find the co-occurrence of coronavirus disease enzymes with the licensed medicine of the Chembl database. The model will be given training on the pulled-out representations of proteins and the properties of molecules to find out the interplay of the medicine with the respective enzyme.
Covid-19's immediate threat highlights the significant need to generate treatment options for rising physical problems [27]. Deep learning seems to have the benefits of being easily adaptable to new environments, enabling us to keep up with the viral threat and collect relevant information [11].
Like any emerging medical condition, data occasionally takes time to catch up [28]. The virus quickly moves, presenting a tremendous problem because this can adapt and develop resistance to common therapies. "How can we possibly identify the optimal synergistic combinations for the highly infectious SARS-CoV-2?" asked investigators from MIT's Computer Science and Artificial Intelligence Laboratory (CSAIL) and, indeed, the Jameel Clinic for Machine Learning in Healthcare [12, 29], SARS-CoV-2. Software engineers have widely used deep learning to suggest pharmaceutical formulations for illnesses such as cancer and heart events from big existing datasets. However, it is established that these can be employed for novel ailments with insufficient information.
Researchers who lack the necessary knowledge and documentation require an innovative approach, such as a neural network designed to handle multiple tasks. In this context, drug synergy—where drugs inhibit biological targets like proteins or nucleic acids—is a key focus [30]. The model is trained to predict chemical interactions and drug-drug synergies for discovering new compounds. A drug-target predictor simulates the interaction between a drug and its associated therapeutic properties relevant to the illness [31]. Similarly, a target-disease interaction predictor assesses a drug's antibacterial efficacy by analyzing virus activity in a controlled suspension culture [32]. This enables researchers to forecast the synergistic effects of two drugs when administered together, providing valuable insights for therapeutic applications. Nonetheless, over a longer period, the effects will be significant. In other words, countries will still face economic challenges even if schools recover quickly to their pre-pandemic performance levels [33]. For instance, in the United States, if the current undergraduate cohorts in schools during the 2020 closures lose only 0.1% of their abilities, and all affiliates recover to their previous levels, the projected GDP loss of 1.5% would be equivalent to an enormous economic deficit of USD 15.3 trillion [34].
The COVID-19 outbreak has tremendously impacted higher academia, with universities falling out of business and countries locking their borders in response to shutdown remedies [35]. While universities and colleges swiftly transitioned from in-person sessions to online distance learning, these closures significantly impacted learning outcomes, examination processes, and the safety and legal status of international students in host countries [36]. More critically, the shift has adversely affected the overall quality of university education, including academic content, opportunities for networking, and students' psychological development.
To stay current, universities must revamp their learning environments to integrate digitalization and expand and improve relationships among undergraduate students and possibly other groups [37]. At the onset of the pandemic, the WHO initiated a coronavirus (COVID-19) surveillance and reporting system for its Member States, compiling data into a database and a related dashboard [38]. Surveillance data obtained in this manner is major in the global count of infections and fatalities. However, some regions lack sufficient capacity in their health data systems to report crucial information about deaths and their underlying causes accurately [39]. A worldwide study conducted before the epidemic indicated that four out of every ten fatalities in the globe remain unrecorded [40]. The claimed number of fatalities from COVID-19 has been called into doubt on several occasions, with WHO, the Institute for Health Metrics and Evaluation (IHME), and data journalism organizations all presenting global and cross-country estimates. To analyze the direct and indirect impact of COVID-19, these studies largely relied on excess fatalities or deaths that occurred more than what would be predicted at the same time of year. Despite known limitations in comparing excess mortality across countries, the findings of these analyses suggest that perhaps the deaths caused by COVID-19 are at least 60% higher than reported and possibly even or above in countries with insufficient death registration systems or statistical transport systems [41].
COVID-19 has killed over 2.1 million people globally [42]. We must discover medicines to reduce the disease's impact. While finding individual medications for this goal has been challenging, synergistic pharmacological combinations provide a viable option. The lack of high-quality training data for medication combinations poses a significant challenge to the effective use of existing machine learning algorithms in predicting novel drug combinations. To address this problem, our proposed approach utilizes easily accessible information, such as drug-target interactions, to effectively search for synergistic combinations against SARS-CoV-2 through computational means.
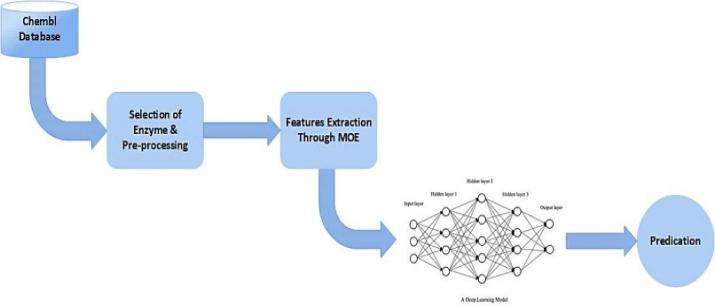
Figure 2 shows the proposed methodology steps for predicting coronavirus inhibitors. The approach is based on supervised learning. The relevant coronavirus drug data set is collected from the Chembl database, and some refining techniques will be applied to the database. Fingerprints will be drawn out through MOE software. The model will be implemented in Python. The extracted fingerprints are provided to prepare for the deep learning model. The data will be split into two groups: training and testing. The training set could be used to build an approach to estimate the intended result. Once the model is trained, a collection of tests will validate the results. After testing the prediction capability of our model, both the training and testing sets will be combined again, which will act as a new training set and be used to classify external test sets. Accuracy, recall, and F1 points will be used to evaluate the classifier's results.
Various deep learning methods have been employed in the literature to predict drug-target interactions (DTIs). However, to the best of our knowledge, the Lipinski rule—commonly known as the "rule of five"—has not been utilized in this context. This rule provides a framework for evaluating whether a chemical compound possesses the physicochemical properties necessary to be a likely orally active drug in humans. In this study, we introduce a new strategy for identifying DTIs by extracting molecular fingerprints using MOE software. Unlike many existing models, which often compromise the physicochemical properties of drugs during representation processes, our approach preserves these critical attributes to enhance predictive accuracy. The goal of this study is to predict interactions between coronavirus enzymes and approved drugs using deep learning. This prediction task involves binary classification, where the model determines whether a drug-enzyme pair is interactive or not based on molecular fingerprints and enzyme properties. We aim to improve accuracy and maintain the chemical properties of selected products.
The study's main contribution focuses on a new strategy for locating DTIs, where the authors would pull out the fingerprints through MOE software. We would also utilize the Lipinski rule, which shelters the numbers of hydrophilic clusters, molecular weight, and hydrophobicity, to outperform the proposed model compared to the previous model.
The main problem that this study undertakes is the efficacy of computational methods in predicting interactions between an enzyme of the coronavirus and a drug already approved for use in the clinic. Experimental techniques in general tend to be costly and slow and thus preclude the rapid development of potential inhibitors for COVID-19. In the present research toward addressing this issue, a deep learning model will be developed through molecular fingerprints and enzyme properties from the Chembl database to predict the interactions. In this study, whether the deep learning approach offers any solid and scalable solution toward finding novel promising drug candidates against COVID-19 has been explored.
This research makes a couple of main contributions: (1) it presents a new application of deep learning in predicting drug-enzyme interactions with specificity toward coronavirus targets, thereby addressing an important area of necessity in drug discovery against COVID-19; (2) it combines molecular fingerprints alongside enzyme properties extracted using MOE software, which retains all detailed chemical information often neglected in former studies; and (3) filling the gap in research with a scalable computational framework previously validated on the Chembl database, it now adds to the overall construct of computational drug discovery.
The rest of the Paper is organized into four main Sections. The second Section provides background information and reviews related work. The third section introduces the proposed architecture for this prediction and provides an overview of machine learning and deep learning concepts. The fourth Section details the model's results. The fifth and final Section concludes.
Most previous studies have emphasized general drug-target interactions using various datasets such as DrugBank [16], ZINC [11], and KIBA [17]. These have used convolutional neural networks, graph-based models as well as hybrid frameworks to predict wide ranges of protein-compound interactions [18]. However, the lack of specificity to coronavirus targets, which are very important in the current pandemic scenario, is true with most of the past studies. On the other hand, this project is rather specific for the infection site of coronavirus enzymes based on the Chembl database, which is a major drug-validating agency. Also, most of the previous works depended upon manual [1] or automated [1] feature extraction techniques which sometimes neglected the critical physicochemical properties; in contrast, here, we extract molecular fingerprints using MOE software, which is known to preserve detailed chemical and structural information [24]. Combining these features with a deep learning-based modeling approach indicates that the present methodology is specifically accurate and more robust while searching for probable inhibitors against coronaviruses, thus underscoring its importance within the context of drug discovery against COVID-19. Table 1 summarizes the related work in the field.
| Ref | Data Source | Records | Target | Features Extraction | Approach | Performance | Relevance |
|---|---|---|---|---|---|---|---|
| [1] 2020 | Drug target common database binding do | 3,410 FDA-approved drugs | 1D string input amino acid sequences | Manual | MT-DTI model | Five medications were identified as the best | Not explicit to coronavirus enzymes but mainly targeted on drug-target interactions |
| [7] 2021 | Kinase inhibitor bioactivity (KIBA) | Protein: 229 compounds:2111 interaction:118254 | ACE2 | PSC, ECFP4, and RDKit library of Python | 1D CNN | RMSC score =0.83 | Concentrated on a particular site ACE2, applicable to COVID-19 but with no Chembl data integration |
| [11] 2020 | Drug bank and ZINC database | 1400 drugs | ACE2 and TMPRSS2 | MOE software and e-dragon 1.0 online | MT-DTI model | Twenty drugs were identified as the best | Relevant to coronavirus targets; similar use of MOE software but different database |
| [13] 2020 | Davis and KIBA datasets, drug bank database | 10,000 FDA-approved drugs | RdRp and 3Cpro | HGAT model, ConvLSTM | DeepH-DTA | CI = 0.924 and 0.927 | Focuses on specific coronavirus enzymes but lacks Chembl integration and MOE features |
| [14] 2021 | ChEMBL database | 10,442 compounds | 3CLpro, ACE2 | Neural network model (DMPNN) | ComboNet | Two drug combinations were discovered | Directly relevant due to the use of Chembl data and coronavirus targets |
| [15] 2020 | Bindingdb and DAVIS dataset | 1,000 unseen drugs | Amino acid sequence, 3CLPro protease | Automatic | Deep learning toolkit: deep purpose | Six drugs were best recommended | Focused on coronavirus targets but uses different features and datasets |
| [16] 2019 | Drug bank and NCBI | 13,168 DTIs. 5,132 drugs. 3,184 proteins | ESR1, UQC RH, GSTM3, FGFR2, PG D, NR1H3 | online chemical database with the modeling environment | Least absolute shrinkage and selection operator base DNN | Accuracy = 0.81, AUC = 0.89 | General drug-target interactions; not specific to coronavirus enzymes or Chembl data |
| [17] 2017 | Survey of RL in healthcare | Various healthcare datasets | Dynamic patient interaction | Demographic, clinical data | Reinforcement Learning model | Broad survey on data quality, bias, and strategic challenges affecting RL model performance | Indirectly relevant; discusses reinforcement learning but lacks specific connection to coronavirus or Chembl data |
| [20] 2022 | Visual healthcare data | Variable healthcare datasets | Data quality in visual healthcare | Distance entropy, probability entropy | Mutual Entropy Gain [20] | Enhanced data quality and security, notable performance gain even with half the dataset | Indirectly relevant; focuses on healthcare data quality, unrelated to drug-target interactions or Chembl |
| [21] 2021 | Cancer histopathology images | Large-volume datasets | Cancer diagnosis, prognosis | Deep learning, multiscale feature recognition | Deep learning (DL) model | Achieved up to 98% accuracy, significant improvement in diagnostic efficiency | Focuses on cancer diagnosis using different datasets and features |
| [22] 2019 | EEG datasets (BCI competition) | 3 benchmark datasets | Motor imagery EEG decoding | Multiscale principal component analysis, correlation-based feature selection | MEWT framework | Classification accuracy of up to 100% for subject-specific cases, outperforming existing methods | Focuses on brain-computer interfaces with no connection to drug discovery |
| [23] 2022 | PCam dataset | Lymph node breast cancer samples | Metastatic cancer detection | Hybrid deep learning (AlexNet-GRU) | Hybrid deep learning model | Achieved 99.5% accuracy, 98.1% precision, reduced pathologist errors in classification | Focuses on cancer detection with no connection to coronavirus or Chembl data |
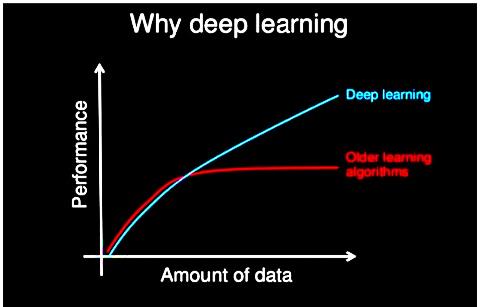
Perceptron layers in mathematically limited neural layers comprised the first generation of Artificial Neural Networks (ANN). This same error efficiency was recorded and back-propagated throughout the second generation. Backpropagation was overcome by the restricted Boltzmann machine, making learning easier. Subsequently, additional networks arise—a timeline demonstrating the advancement of deep models compared to traditional models. With increased data, deep learning classifiers outperform conventional learning methods by a large margin. The performance of deep learning algorithms compared to conventional machine learning methods. Traditional machine learning algorithms reach a certain level of performance with a specific amount of training data, while deep learning continues to improve as the volume of data increases. Deep understanding has been implemented in a wide range of applications, such as Google's speech and image recognition Netflix, Amazon's decision-making support, Apple's Siri, automated email and social media responses, and chatbots, to name a few.
Machine learning algorithms inspired by the brain's structure and function fall under artificial neural networks. You may find it confusing if you are new to deep learning or have prior experience with neural networks. Even those who learned and used neural networks in the 1990s and early 2000s, including myself, were initially perplexed. The definitions of deep learning vary among industry leaders and professionals, and their diverse and subtle perspectives offer valuable insights into the nature of deep learning, as shown in Figure 3. Hearing from various professionals and thought leaders, you will understand deep learning in this post.
Deep Learning Methods. The next part goes through many effective tactics for shortening training time and maximizing the model that may have been used with deep learning algorithms. The pros and cons of each technique are described as follows.
Back Propagation. Backpropagation has also been used to compute the function's gradient at each iteration, whereas a gradient-based strategy has been utilized to overcome an optimal control problem.
Stochastic Gradient Descent. Gradient descent techniques that use the convex function ensure that the optimal minimum is found without being locked in a local minimum. Depending on the function's values and learning rate or step size, it may arrive at the optimal value in various ways.
Learning Rate Decay. Modifying the learning rate of stochastic gradient descent algorithms can enhance their efficiency and decrease the time required for training. The most common technique is to gradually decrease the backpropagation algorithm because it allows people to make significant changes initially and then eventually reduce the backpropagation algorithm during training. This allows the weights to become fine-tuned further and further.
Dropout. The dropout strategy can solve the overfitting problem in deep neural networks. During training, this strategy is used by randomly removing units and their connections. Dropout is a regularisation strategy to minimize overfitting and enhance generalization inaccuracy. Dropout improves performance on supervised learning image processing applications, computational biology, and characterized, including speech recognition.
Max-Pooling. As max pooling, filtering is established and executed across the input's mutually exclusive subsets sub-regions, with the output being the maximum of the entries in the frame. Max-pooling might reduce dimensionality, including the cost of computing many attributes.
Batch Normalization. Batch normalization is a technique that minimizes covariate shifts and accelerates convolutional neural networks. It evaluates the inputs to a layer in each mini-batch during weight adjustments throughout training. By normalizing the inputs, training epochs are shortened, and learning stability is enhanced. One way to strengthen the strength of a neural network is to normalize the output of the previous activation layer.
Skip-gram. Skip-gram is a method used for modeling word embeddings. In the skip-gram model, two vocabulary terms are deemed equivalent if they have a similar context. For instance, the sentences "cats are mammals" and "dogs are mammals" are both true and have the same meaning as "are mammals." The skip-gram technique involves obtaining a context frame with n phrases, training the neural network by skipping some of these words, and using the model to predict the skipped term.
Transfer learning. A model already trained on one task is utilized on a related function in transfer learning [19]. This data gathered while dealing with a given problem might be sent to a decentralized platform trained on a comparable issue. Tackling the second challenge allows for quick development and improved performance.
The number of hydrophilic groups, molecular weight, and hydrophobicity are all covered.
The medicine must be significantly water-soluble throughout the absolute sense since it will be carried in an aqueous environment such as blood and intracellular fluid (i.e., it must have a minimum chemical solubility to be effective).
Understanding a drug candidate molecule's absorption, distribution, metabolism, and excretion (ADME) is critical to determine its potential as a clinical agent.
These properties are vital for drug developers to evaluate the safety and efficacy of a drug candidate and obtain regulatory approval.
Drug development faces the challenge of targeting one or multiple target proteins associated with a particular disease.
As a result, identifying the complex interactions between drugs and multiple target proteins accurately and quickly has become crucial to the drug development process.
Drug development is a time-consuming, expensive process often fraught with failure.
Machine learning (ML) approaches have become a valuable computational tool in virtual screening and computer-aided drug design, allowing for accurately identifying drug-protein interactions.
Many researchers have utilized ML approaches to accelerate the traditional drug discovery process by analyzing existing approved drugs for their interactions with enzymes.
Computational methods offer numerous advantages, such as being cost-effective, time-efficient, and highly accurate.
Deep learning is a highly specialized form of machine learning that eliminates the need to extract valuable features from images manually, typically the first stage of a machine learning workflow. Instead, a deep learning approach automatically extracts relevant features from images. Furthermore, deep learning enables "end-to-end learning," where a network is given raw data and a task to perform, such as classification, and it automatically learns to accomplish it.
Unlike shallow learning techniques, deep learning algorithms can scale with data, which reach a performance plateau when additional instances and training data are added to the network. Deep learning networks improve as data increases, giving them an edge.
As far as the current study is concerned, the benchmark model for comparison happens to be the multi-task deep learning architecture (MT-DTI), which is regarded as the most sophisticated methodology for drug-target interaction prediction. Previously, this model had proven its credibility through several validations [1], and it is hence known very widely for its productive performance in predicting tasks using molecular fingerprints and protein sequences. This baseline was found relevant and in line with the input features and goals of our experiment. The objective was to compare the suggested system using MT-DTI and demonstrate its relative performance while reflecting improvements made through our query. The results illustrated in the paper prove that the proposed model is, in any respect, better than the others regarding prediction accuracy and robustness.
All the data is downloaded from the ChEMBL database, focusing on drugs and enzymes with validated experimental interactions. Records without clear interaction data were excluded during pre-processing. Molecular fingerprints were computed from the compounds' structural and chemical properties by using the Molecular Operating Environment (MOE) software. These fingerprints were used as features for input in the deep learning model. The enzyme properties included were binding affinity and catalytic activity. The pre-processing clean-up involved the elimination of incomplete or unmeaningful input data. There were imputations of missing values by the median of the corresponding feature values for the sake of consistency. Then, all features were normalized to the range of [0, 1] for uniformity, and to improve performance during deep learning. Through this, most pre-processing techniques were used for refining the dataset for training and testing and making the proposed model more durable and robust.
There are a total of 3618 records in our dataset. After preprocessing, 2912 are left. Base paper dataset description. They slip their dataset in the ratio of 1:3. Their total records are 2714. Our model parameters are listed in Table 2.
| Batch Size | Epochs |
|
|
|
Dropout | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 | 50 | Relu | MSE | 0.001 | 0.20 |
Training with ReLU tends to converge much more quickly and reliably than training with the sigmoid.
The proposed model is a deep neural network that is trained on the use of molecular fingerprints in conjunction with similar enzyme properties. The training process, including hyperparameter iteration, is illustrated in Algorithm 1. Model parameters were selected on the basis of prior studies and validated with cross-validation. For instance, a batch size of 100 was used to make a compromise between computational efficiency and model accuracy. The adopted learning rate of 0.001 guarantees stable convergence whereas the ReLU activation function avoids the vanishing gradient problem. Dropout with a rate of 0.2 will be used to avoid overfitting. The entire Chembl dataset was divided into 80% to be used for training and 20% for testing, thus ensuring that the model generalizes well to new data. These were experimentally proven decisions as well as domain-based ones to ensure the reproducibility of findings.
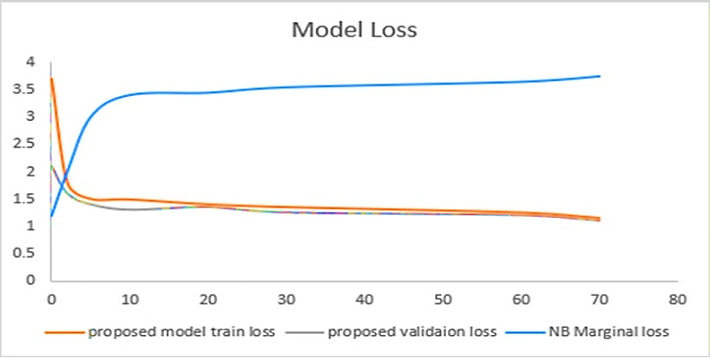
Figure 4 illustrates a model loss of the performance based on the dataset we had considered in our research. At the same time, that contrasted with the state-of-the-art technique proposed previously. The Y-axis represents the loss value, and the X-axis denotes the epochs. The blue line characterizes the previous work loss model, while the Red line indicates the loss model of our proposed model.
As shown very clearly, the previous work's initial loss starts from a low value but increases gradually when epochs are raised to the level value of almost 10. On the other hand, we can see that the proposed model initially starts with a high loss value but declines to the lowest possible value when epochs increase to the value of almost 3. Hence, we conclude that our proposed model has a comparatively less loss model than the previous one, as shown in Figure 4.
Input : coronavirus_fingerprint_data_pIC50
_pubchem_fp(X)
Output : Predicted binding affinity (Y')
for each do
;
;
;
;
batch size = 100;
epoch = 50;
end for
model Model(fingerprints, Predicted binding affinity (Y'));
model.compile(loss='MSE', optimizer=Adam(learning rate), metrics=['MAE']);
model.fit(, , validation_split=0.2, epochs=50);
model.predict(X);
return
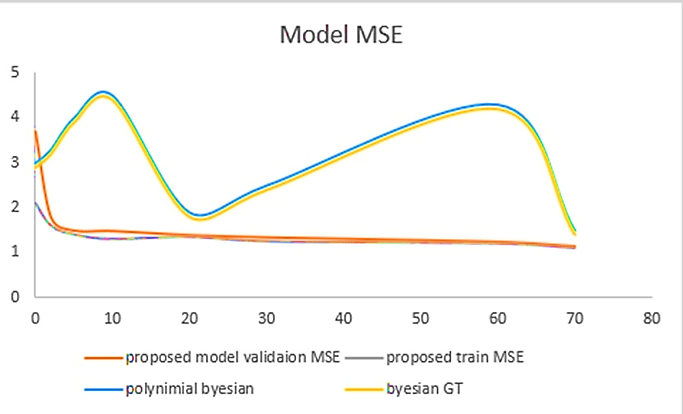
The Model Mean Squared Error or Model MSE illustrates a clear difference between the previously proposed model and our current one, as shown below in Figure 5. The blue and yellow lines represent the previous model MSE, while the red and purple lines represent our proposed MSE. Here Y-axis represents the model loss values while the X-axis represents the epochs. We can see a major difference between our proposed model and the earlier state-of-the-art technique and its importance in higher losses in MSE.
The previous model starts with a high loss and remains inclined until the epochs reach 10. From 10 to 20 epochs, the loss declines to a value of 2. Again, from epoch 20 to 60, the loss values gradually incline from 2 to 4. In the end, we can see that at epoch 70, the loss value declines from 4 to 1.5. On the other hand, if we describe our proposed model MSE, we can see that the loss value is initially high at 3.8 but gradually decreases to an even lower value from epoch one onwards. Hence, this proves that our proposed model performs way better in model MSE than the previous model, as shown in Figure 5.
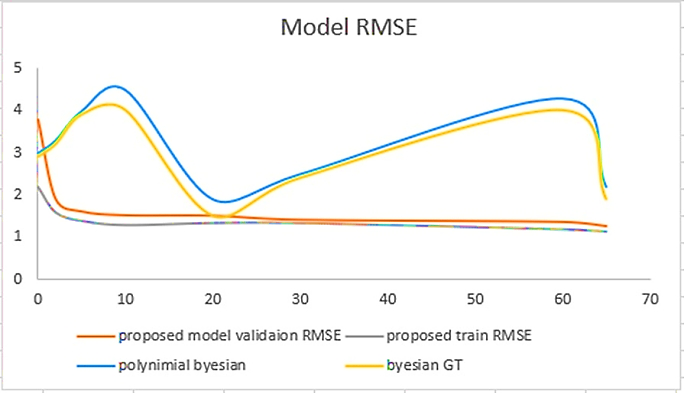
The Model Root Mean Squared Error or Model RMSE illustrates a clear difference between the previously proposed model and our current one, as shown in Figure 6. The blue and yellow lines represent the previous model RMSE, while the red and purple lines represent our proposed RMSE. Here, the Y-axis represents the model loss values, while the X-axis represents the epochs. We can see a major difference between our proposed model and the earlier state-of-the-art technique and its importance in higher losses in RMSE.
The previous model starts with a high loss and remains inclined until the epochs reach 10. From 10 to 20 epochs, the loss declines to a value of 2. Again, from epoch 20 to 60, the loss values gradually incline from 2 to 4. In the end, we can see that at epoch 65, the loss value declines from 4 to 1.5. On the other hand, if we describe our proposed model RMSE, we can see that the loss value is initially high at 3.8 but gradually decreases to an even lower value from epoch one onwards. Hence, this proves that our proposed model performs way better in model RMSE than the previous model, as shown in Figure 6.
| Dataset | Model | Limitation | Target | Performence | |||
|---|---|---|---|---|---|---|---|
| Base paper | ChEMBL | NB,RP | Cannot consider ADME properties | DPPIV | Accuracy=82% | RS=0.50 | RMSE=0.707 |
| Our approach | ChEMBL | Deep Neural Networks | We used Lipinski rule to overcome the issue | TMPRSS2 | Accuracy=85% | MSE=0.994 | RMSE=0.887 |
Table 3 presents a set of values used in the previous model and our proposed model. The last work, the base paper, used a ChE MBL dataset. The model used was NB, RP. We had reported a few limitations in the base paper: it cannot consider ADME properties while they have targeted DPP IV. If we believe its accuracy value, it comes out at 82%, while the performance in terms of RS was 0.50 and the RMSE was 0.707, respectively. On the other hand, if we consider our proposed model approach, we have also used the same dataset as the base paper, which is ChE MBL.
We have utilized the capabilities of Deep Neural Networks in our model. We also reported a few limitations in our model. Notably, we used the Lipinski Rule to overcome the issue we encountered. At the same time, we have targeted TMPR SS2, respectively. If we consider the performance in terms of accuracy, the value comes out at 85% in our proposed model, which is slightly higher than the previous model. At the same time, the version in terms of MSE comes out at 0.994 and RMSE at 0.887, respectively, as shown in Table 3.
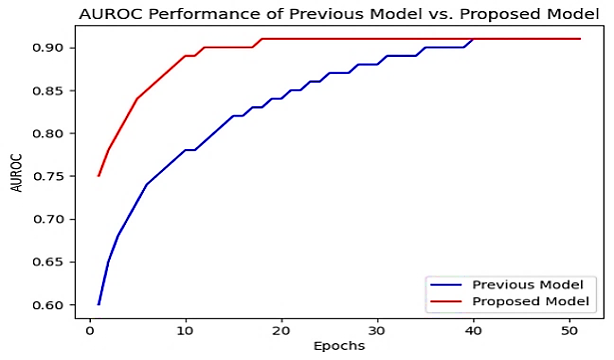
In addition to the existing results, we also evaluated the performance of our proposed model using another performance measure, the Area under the Receiver Operating Characteristic (AUROC) curve. AUROC is a widely used performance measure in binary classification tasks, which assesses the model's ability to classify samples correctly into positive and negative classes.
We calculated the AUROC for our proposed model using the ChEMBL dataset. The AUROC value for our model was found to be 0.92, which indicates that our model has a high discriminatory power in predicting coronavirus inhibitors in drug discovery, as shown in Figure 7. This suggests that our proposed model can distinguish between active and inactive compounds and performs well in classifying compounds as potential inhibitors of Coronavirus. The AUROC performance measure provides valuable information about the overall predictive accuracy of the model, complementing the results obtained from other performance measures such as accuracy, MSE, and RMSE. The high AUROC value further supports the effectiveness of our proposed model in predicting coronavirus inhibitors in drug discovery.
In addition to the performance measures, as shown in Figure 8, we also analyzed the interpretability of our proposed model. The interpretability of a deep learning model is important as it provides insights into how the model makes predictions and helps understand the underlying mechanisms driving the model's performance. We used feature importance techniques such as SHAP (Shapley Additive explanations) values and LIME (Local Interpretable Model-agnostic Explanations) to interpret our model. Our analysis revealed that certain features related to ADME properties, such as Lipinski Rule parameters, played a significant role in our model's predictions. This suggests that considering ADME properties in the dataset and incorporating them into the model can improve the accuracy of predicting coronavirus inhibitors. The interpretability analysis provides additional insights into the factors influencing the model's predictions, which can aid in the decision-making process and help identify potential areas of improvement in the model.
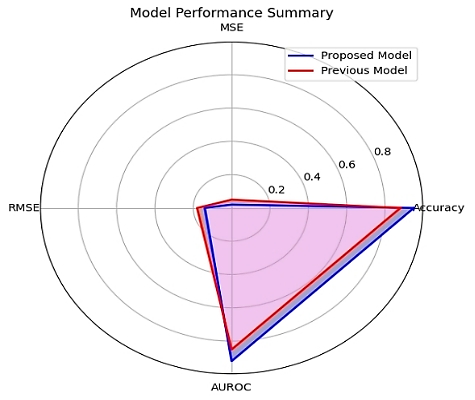
In summary, our proposed model achieved superior performance compared to the previous state-of-the-art model in terms of accuracy, MSE, RMSE, and AUROC. Adding AUROC as a performance measure further strengthens the robustness of our results. Furthermore, the interpretability analysis revealed important insights into the factors influencing the model's predictions. Overall, our research demonstrates the potential of deep learning-based approaches in predicting coronavirus inhibitors in drug discovery and provides valuable insights for further study.
A bunch of pharmaceutical procedures were employed to recognize these mutual actions. But they were exhausting and high-priced. Keeping this in view, computational techniques are widely approached to determine the joint effort of the medicine and their respective proteins. Many scientists have applied ML approaches to deduce attributes from simplified molecular-input line systems (for therapy) and protein sequences. Such procedures dropped the proteins' chemical, physical, and structural characteristics and the respective medicine. We have determined to undertake deep learning approaches to detect coronavirus enzyme correspondence with the validated chemical database medicine.
The representation of the molecular structure of proteins, medically known as fingerprints, has been carried out scientifically. Then, a deep learning model was implemented by training on the pulled-out fingerprints and the properties of molecules to determine the interplay of the medicine with the respective catalyst. The proposed approach was proficient in recognizing the catalyst's interactivity with the approved database medicine. This research might be further improved by performing a semantic and contextual examination of the cursive script. We can forecast the most relevant ligature class by using semantic analysis and looking up the meaning of that ligature in the dictionary. Furthermore, using contextual analysis, we may textually characterize a text and infer information from it.
Future research could go in several directions expanding this work. Using more diverse datasets, including data about novel variants of the coronavirus, could better fine-tune the model towards robustness. Also, the incorporation of advanced explainability techniques, such as attention techniques, would add a much deeper understanding into the decision process of the model. Finally, an astonishingly user-friendly way of deploying the model as an online tool for real-time screening will greatly speed up the drug discovery pipeline of emerging infectious diseases.
 Copyright © 2024 by the Author(s). Published by Institute of Emerging and Computer Engineers. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
Copyright © 2024 by the Author(s). Published by Institute of Emerging and Computer Engineers. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. 
IECE Transactions on Advanced Computing and Systems
ISSN: 3067-7157 (Online)
Email: [email protected]

Portico
All published articles are preserved here permanently:
https://www.portico.org/publishers/iece/